2012年4月6日作成
2013年6月2日最終更新(genotypes と Coverage depth に関する記述を増しました)
注)本ホームページの情報は古く、最近のバージョンには対応していません。
しかし、考え方は基本的に同じなので、参考のため残しています。最近のバージョンの使い方は、stacksのホームページをご覧ください。「フリーソフトを使った QTL 解析」の最初の部分も、参考になると思います。
はじめに
重要!
サンプルファイル
使用環境
Stacks をセットアップする
SNPs 探索の準備
SNPs の探索
1.集団解析
2.親 n 個体から作成した交配家系を用いたマッピング
出力ファイルの見かた
並列処理
Coverage depth
最後に
引用
はじめに
非モデル生物など遺伝情報があまり知られていない生物からでも、大量に SNPs(一塩基多型)を取得できる Restriction site Associated DNA (RAD) sequencing という方法が 2008 年に発表されました[1]。 この方法では、ゲノムを制限酵素で切断し、切断した部位から 50–100 bp 程度のシーケンスを読み、そのシーケンスから SNPs を探索します。 シーケンスには通常、次世代シーケンサーの illumina が使われます。 これまで、飼育家系を用いた連鎖解析[e.g. 2]、野生集団を用いた association study[e.g. 3]、系統地理[e.g. 4]、またこれらを組み合わせた研究[e.g. 5]など、さまざまな研究に応用されています。
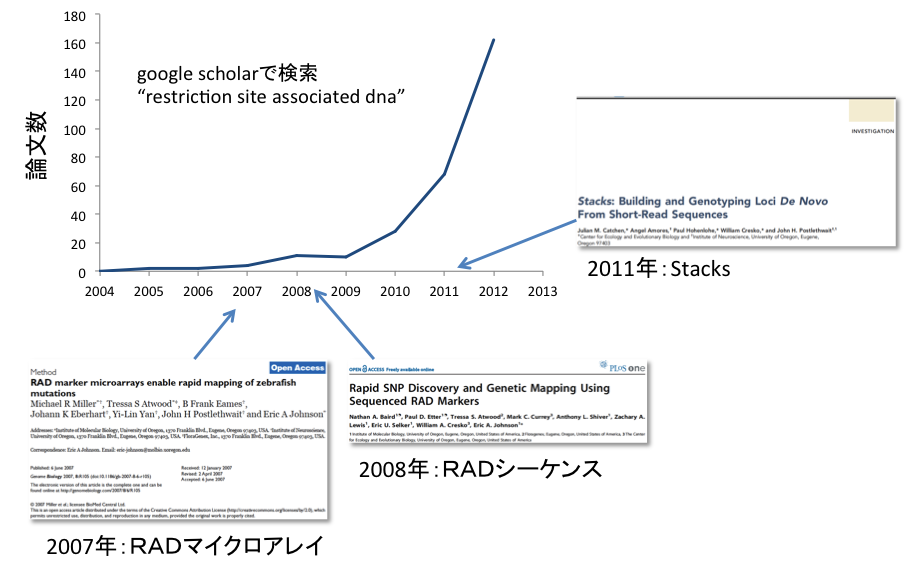
RAD には幾つかの方法があります。 例えば一つの III 型制限酵素を使う方法[1,2,4,5]と、IIB 型制限酵素を使う方法[6]があります。後者ではゲノムの物理的断片化(shearing、高価な専用の機材が必要)を行わなくてもよいという利点がありますが、一般にデータ量が少なくなると考えられます。 二つの III 型制限酵素を使う事により、ゲノムの物理的断片化を行わない方法も行われています[3]。 またシーケンスには、single read[1,3,4] (DNA 断片を制限領域からだけ読む)を使った方法だけでなく、paired-end[2,5,7] (DNA 断片を制限領域とその反対側の両方から読む)を使った方法も行われています。 この文章では、もっともシンプルな方法、つまり一つの III 型制限酵素で切断し、single read でシーケンスした場合を例に説明します。
ソフトウェア Stacks[8]は、illumina などから得られた膨大な RAD シーケンスデータから SNPs を見付けるのに威力を発揮します。 Stacks の機能を 100% 活用するには、コンピューターに AMP 環境(Apache, MySQL, PHP)を構築する必要があります。しかし、これは結構大変です。ここでは、AMP 環境を使わずに Stacks を使う方法を紹介します。
説明はできるだけ分かり易く、UNIX や Linux を使ったことのない人でも分かるように心がけました。RAD は理解していることを前提としています。
なお、分かり易くするため、細かい説明は省きました。また、厳密には正確ではない部分もあります。 Stacks の詳細、および正確な説明は、Catchen et al.[8] をご覧下さい。
重要!
当ホームページによって生じた不利益につきましては、一切の責任を負いかねますのでご了承ください。
ご紹介する方法は、Stacks-0.999991 (updated May 15, 2013) で使えることを確認しています。
サンプルファイル
下はサンプルファイルです。 ファイルサイズが小さいので、コンピューターのメモリが小さくても最後まで計算できます。 ZIP で圧縮しているので、解凍してお使い下さい。 「SNPs 探索の準備」に従って、所定のディレクトリに入れてお使いください。 バーコードはAAAAA, CCCCC, GGGGG, TTTTTの4個体分です。
・バーコードのファイル:barcodes.zip (4 KB)
・データファイル:ExampleData.fq.zip (4.3 MB)
使用環境
UNIX または Linux。
当ホームページでは Mac OS X Mountain Lion 10.8.3 (UNIX) を使った場合で説明しています。しかし、Stacks を使う環境を整えるのがやや面倒なうえ(「Stacks をセットアップする」を参照)、パイプライン(プログラム)のひとつである genotypes のインストールが上手く行きませんでした(今回は、過去にダウンロードした古いバージョン Stacks-0.9991 の genotypes を使いました)。Stacks と Mac の相性はあまり良くないのかも知れません。なお、Linux ではこのような問題もなくインストールできました。
メモリは多い方が良いです。パラメーターの設定やデータ量によって異なりますが、illumina hiseq 2000, 1 lane, single read のデータ (約 140 million reads) を解析したときは、32 GB でも足りないことがありました。
必要なハードディスクの空き容量も、解析によって異なります。私の行った解析(illumina hiseq 2000, 1 lane, single read)では、全て含めて 180 GB 程度でした
なお、上で紹介したサンプルファイルはサイズが小さいので、メモリもハードディスクの空き容量もそれほど必要ありません。普通のノートパソコンでも簡単に解析できます。練習用にお使いください。
Stacks をセットアップする
Mac OS X Mountain Lion ではまず、Xcode、Command Line Tools (OS X Mountain Lion) for Xcode、そして MacPorts をインストールします。それぞれ、利用可能なバージョンのうち最新のものをインストールして下さい。Xcodeは、インストールしたらアイコンをクリックして、一度立ち上げてみてください。なお Xcode はファイルサイズが大きく、ダウンロードに時間がかかります(4.6.2.dmg では 1.61 GB)。
下の URL から Stacks をダウンロードします。
http://creskolab.uoregon.edu/stacks/
stacks-xxxxx.tar.gz をダブルクリックして解凍します。xxxxx はバージョンの番号です。以降の説明やコマンドで stacks-xxxxx となっている部分は、お使いのバージョンによって適宜変更してください。
解凍したフォルダ stacks-xxxxx を「アプリケーション」フォルダに入れます。入れる場所はどこでも構いませんが、ここでは便宜的に「アプリケーション」フォルダに入れることにします。
ターミナルを開きます(「アプリケーション」>「ユーティリティ」>「ターミナル」)。
カレントディレクトリを stacks-xxxxx にします。
|
~ username$ cd /Applications/stacks-xxxxx stacks-xxxxx username$ |
下の2つのコマンドを実行します。
|
stacks-xxxxx username$ ./configure stacks-xxxxx username$ make |
SNPs 探索の準備
データや解析結果を入れるフォルダを作ります。ここでは、ホームディレクトリに tutorial というフォルダを作ります。
|
stacks-xxxxx username$ mkdir ~/tutorial |
注)Mac の Finder 上でフォルダを作っても OK です。
フォルダ tutorial のなかに3つのフォルダ raw, samples, stacks を作ります。それぞれには生データ、加工したデータ、解析結果が入ります。
|
stacks-xxxxx username$ mkdir ~/tutorial/raw stacks-xxxxx username$ mkdir ~/tutorial/samples stacks-xxxxx username$ mkdir ~/tutorial/stacks |
注)フォルダ stacks-xxxxx とフォルダ stacks は異なるので、混同しないように気をつけてください。
テキストエディタを使って、RAD に用いたバーコードのファイルを作ります。改行コードは必ず LF (UNIX) にしてください。フリーソフトの mi でできます。ファイル名を barcodes にして、フォルダ tutorial に保存します。バーコードのサンプルファイル (4 KB) はこちらです。
|
AAAAA(改行) CCCCC(改行) GGGGG(改行) TTTTT(改行) などなど、以下省略 |
データファイル(1つ、もしくは複数)を全てフォルダ raw に入れます(Finder 上でドラッグ&ドロップ)。圧縮してある場合は、解凍してから入れてください。くれぐれも、圧縮ファイルをこのフォルダに入れないで下さい。フォルダ raw には、解凍したデータファイルのみが入るようにして下さい。データファイルのサンプル (4.3 MB) はこちらです。
フォルダの構成は、下のようになっているはずです。
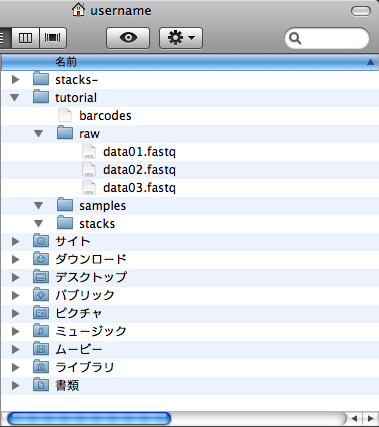
(図中の "stacks-" は無視して下さい)
SNPs の探索
調べる内容によって、探索方法が2つあります。
1.集団解析:全個体から SNPs を探索し、遺伝子型を調べます。
2.親 n 個体から作成した交配家系を用いたマッピング:親 n 個体から SNPs を探索し、得られた SNPs について F2 などマッピングに使う個体の遺伝子型を調べます。
1.集団解析
普通は多くのサンプルを同時に解析しますが、4サンプルを例に説明します。1サンプルに複数個体を混ぜて解析することもありますが[e.g. 4]、ここでは1サンプル1個体とし、各個体に固有の RAD タグ(AAAAA, CCCCC, GGGGG, TTTTT)をつけた場合で説明します。
1-1. process_radtags:シーケンスデータを個体ごと(RAD タグごと)のファイルに分け、フォルダ samples に保存します。
|
tutorial username$ /Applications/stacks-xxxxx/process_radtags -p ./raw/ -o ./samples/ -b ./barcodes -c -r -e ecoRI -t 75 -i fastq |
-c オプションを指定すると、uncalled base (つまり "N")の含まれたリードが除外されます。
-e オプションでは、RAD sequencing に使った制限酵素(sbfI, pstI, ecoRI, sgrAI のいずれか)を指定します。
-t オプションでは、解析に用いる配列の長さを指定します。生データが 100 bp であっても、80 塩基目よりあとの信頼性が低い場合、81〜100 塩基目を解析から除外することができます。そのときは、頭のバーコード(今回の例では 5 bp)を除いた 6〜80 塩基目の 75 bp について解析することから、-t 75 と指定します。
-i オプションでは、データファイルの保存形式を指定します(fastq または bustard)。
他のオプションについては、Stacks のホームページにある process_radtags の説明を参照してください。
注) 久しぶりに最新のバージョン(ver 1.35)をダウンロードしてみたら、どんな拡張子でも解析してくれる訳ではありませんでした。少なくとも .fq と .fastq では解析してくれましたが、他の拡張子は試していません。(2015/10/02追記)
Tips)このパイプラインでは、フォルダ raw 内の全てのファイルを解析します。このため、フォルダ raw に別のファイルがあると、解析が上手く行きません。例えば Mac では、フォルダ raw に .DS_Store という隠しファイルが入っていることがあります。そうすると、下のようなエラーが出て解析できません。この場合は、コマンド rm ./raw/.DS_Store を実行して .DS_Store を削除してから解析して下さい。
|
Found 4 input file(s). Loaded 4, 5bp barcodes. Processing file 1 of 4 [.DS_Store] Unable to allocate Seq object. |
Tips)解析が上手く行かない場合は、データファイルを一つにまとめて、-f オプションを使ってファイル名を明示的に指定すると上手く行くことがあります。下の例では、data01.fastq, data02.fastq, data03.fastq という3つのデータファイルをひとつにまとめて、data.fastq というファイルにして解析しています。
|
(データファイルを一つにするコマンド↓) tutorial username$ cat raw/data01.fastq raw/data02.fastq raw/data03.fastq > raw/data.fastq (解析を実行するコマンド↓) tutorial username$ /Applications/stacks-xxxxx/process_radtags -f ./raw/data.fastq -o ./samples/ -b ./barcodes -c -r -e ecoRI -t 75 -i fastq |
確認)ここでは、フォルダ samples に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls samples process_radtags.log sample_AAAAA.fq sample_CCCCC.fq sample_GGGGG.fq sample_TTTTT.fq |
1-2. ustacks:個体ごとに、全く同じ配列を持ったリードを束ねて stack とします。また stacks 同士を比較して、個体内で同じ遺伝子座(RAD locus)のものをひとまとめにし、SNPs を検出します。今回の例では、ヘテロの SNPs (エラーによる、アリルが3つ以上の SNPs も)を検出することになります。
|
tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_AAAAA.fq -o ./stacks -i 1 -d -r -m 3 -M 2 -p 4 tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_CCCCC.fq -o ./stacks -i 2 -d -r -m 3 -M 2 -p 4 tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_GGGGG.fq -o ./stacks -i 3 -d -r -m 3 -M 2 -p 4 tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_TTTTT.fq -o ./stacks -i 4 -d -r -m 3 -M 2 -p 4 |
-i オプションには、個体ごとに異なる値を指定します。
-m オプションは、stack として認めるのに必要なリードの本数を指定します。個体内では、全く同じ配列を持ったリードのグループが多数存在します。このうち、リードの本数が -m の値以上のグループを stack と呼び、解析に供します。いっぽうリードが 1〜2 本しか存在しないマイナーなグループは、エラーによって生成された可能性があります。このようなエラーの影響を小さくするため、リードの本数が -m の値より小さいマイナーなグループは stack とはせず、解析から除外する事もできます。一般にはこのオプションを 3 に設定することが多いようです。しかし、coverage depth(遺伝子座あたりのリード数、下の 「Coverage depth」参照)が非常に大きい場合は、エラーによって全く同じリードが3本以上生成される確率も高まります。このことから、coverage depth が大きい個体では、-m の値を大きくするべきかも知れません。
-M オプションは、個体内で stacks が同じ遺伝子座のものと判断される際に許される塩基の違いです。つまり、2つの stacks 間の距離(異なる塩基の数)が -M の値より小さければ、両者は同じ遺伝子座に分類されます。逆に 2 つの stacks 間の距離が -M の値より大きければ、両者は異なる遺伝子座に分類されます。このため、-M の値を小さく設定しすぎると、本当は同じ遺伝子座の stacks なのに、誤って違う遺伝子座に分類してしまうリスクが高まります。逆にこの値を大きく設定しすぎると、本当は異なる遺伝子座の stacks なのに、誤って同じ遺伝子座に分類してしまうリスクが高まります。このため、適度な値を設定する必要があります。一般には、解析する個体内の遺伝的多様性が高い場合(ヘテロの遺伝子座が多い場合)には大きく、低い場合には小さく設定するのが良いでしょう。経験的には、野生個体では 2 もしくは 3 くらいに設定することが多いようです。
他のオプションについては、Stacks のホームページにある ustacks の説明を参照してください。
Tips)ustacks の複数のコマンド(上の例では AAAAA、CCCCC、GGGGG、TTTTT のコマンド)を一度に入力して解析することが出来ます。この場合、別のソフトでコマンドを作成してコピペすると便利です。しかし、改行が入っていると解析が上手く行きません。コマンドは改行ではなく、セミコロン ; でつなげると上手くいきます。
Tips)コマンドを実行すると、文字がずらずらと出力されます。その中に Mean coverage depth と Mean merged coverage depth が表示されます(両者の違いは Catchen et al.[8]をご覧下さい )。これらは、解析結果の信頼性を評価するのに重要な値です。出力を command + s (Mac の場合)で保存しておくと、あとあと便利です。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks sample_AAAAA.alleles.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
1-3. cstacks:個体間で同じ遺伝子座の stacks をまとめて SNPs を検出し、全個体に見られる遺伝子座のカタログと SNPs のカタログを作ります。
|
tutorial username$ /Applications/stacks-xxxxx/cstacks -b 1 -o ./stacks -s ./stacks/sample_AAAAA -s ./stacks/sample_CCCCC -s ./stacks/sample_GGGGG -s ./stacks/sample_TTTTT -p 4 -n 3 |
-n オプションは、個体間で許されるミスマッチの数を指定します。例えば前の解析(ustacks)において、個体1で stack A と stack B が遺伝子座1に、個体2で stack C と stack D が遺伝子座2に分類されたとします。そして stack A と stack C の距離(異なる塩基の数)を A-C、ほかも同様に A-D、B-C、B-D としたとき、この4つの距離のうち最も小さい値(仮に D とします)が -n 以下であれば、個体間で遺伝子座1と遺伝子座2が相同であると判断されます。逆に D が -n より大きければ、遺伝子座1と遺伝子座2は異なるものであると判断されます。このため -n が小さすぎると、遺伝子座1と遺伝子座2が本当は相同であっても、誤って異なる遺伝子座に分類されるリスクが高まります。逆にこの値が大きすぎると、遺伝子座1と遺伝子座2が本当は異なるのに、誤って相同な遺伝子座であると分類されるリスクが高まります。よって、適切な値を設定する必要があります。一般には、集団解析で個体数が十分に多い場合、0 と設定してもよいようです。この場合、個体1の遺伝子座1と個体2の遺伝子座2で D = 0 であれば(例えばそれぞれの遺伝子型が AC/TC, TC/TG だった場合)、両者は相同と見なされます。また個体2の遺伝子座2と個体3の遺伝子座3でも D = 0 であれば(例えば TC/TG, AG/TG)、それらは相同と見なされます。つまり、遺伝子座 1、遺伝子座 2、そして遺伝子座 3 は全て相同と見なされます。ここで重要なのは、遺伝子座 1 と遺伝子座 3 の間で D = 0 でなくても(上の例では AC/TC と AG/TG の間で D = 1)、相同と見なされることです。解析に用いた個体数が十分に多い場合は、このように相同な遺伝子座が個体間で数珠つなぎ状に連なることが期待できるため、-n を 0 とすることができます。
他のオプションについては、Stacks のホームページにある cstacks の説明を参照してください。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks batch_1.catalog.alleles.tsv batch_1.catalog.snps.tsv batch_1.catalog.tags.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
1-4. sstacks:各個体で、SNPs の遺伝子型を決めます。
|
tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_AAAAA -o ./stacks -p 4 tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_CCCCC -o ./stacks -p 4 tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_GGGGG -o ./stacks -p 4 tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_TTTTT -o ./stacks -p 4 |
オプションについては、Stacks のホームページにある sstacks の説明を参照してください。
Tips)sstacks の複数のコマンド(上の例では AAAAA、CCCCC、GGGGG、TTTTT のコマンド)を一度に入力して解析することが出来ます。この場合、別のソフトでコマンドを作成してコピペすると便利です。しかし、改行が入っていると解析が上手く行きません。コマンドは改行ではなく、セミコロン ; でつなげると上手くいきます。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks batch_1.catalog.alleles.tsv batch_1.catalog.snps.tsv batch_1.catalog.tags.tsv sample_AAAAA.alleles.tsv sample_AAAAA.matches.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.matches.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.matches.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.matches.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
1-5. genotypes:解析のまとめを作ります。全個体の遺伝子型が表になって出力されます。
|
tutorial username$ /Applications/stacks-xxxxx/genotypes -b 1 -P ./stacks -r 1 -c |
-r オプションは、遺伝子座のフィルタリングと関係します。下の表のように、遺伝子座によって遺伝子型が推定された個体と推定されなかった個体があります。たとえば遺伝子座2では、個体 TTTTT で遺伝子型が推定されていません(ヌル)。遺伝子型が推定されなかった要因として coverage depth が低かったり、制限酵素の認識部位の配列が他個体と異なるためシーケンスが読まれなかったなどが考えられます。このため、遺伝子座によって遺伝子型が推定された個体数が異なります(表の右端の列)。-r オプションは、遺伝子型が推定された個体数の多い遺伝子座のみを出力します。例えば -r 3 と指定すると、下の例では、遺伝子型が推定された個体数が3以上の遺伝子座(つまり遺伝子座1と2)が出力ファイルに反映さます。遺伝子型が推定された個体数が3未満の遺伝子座(つまり遺伝子座3と4)は、出力ファイルから除外されます。このとき、出力ファイル batch_1.haplotypes_1.tsv のファイル名の最後の数字が3になります(batch_1.haplotypes_3.tsv)。この値は、Rubin et al. (2012)[9] の min.taxa に相当します。
| 個体 AAAAA | 個体 CCCCC | 個体 GGGGG | 個体 TTTTT | 遺伝子型が推定 された個体数 |
|
| 遺伝子座1 | A/G | A | G | A/G | 4 |
| 遺伝子座2* | CA | TG | CA/TG | - | 3 |
| 遺伝子座3 | - | A | - | G | 2 |
| 遺伝子座4 | C/T | - | - | - | 1 |
他のオプションについては、Stacks のホームページにある genotypes の説明を参照してください。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks batch_1.catalog.alleles.tsv batch_1.catalog.snps.tsv batch_1.catalog.tags.tsv batch_1.haplotypes_1.tsv sample_AAAAA.alleles.tsv sample_AAAAA.matches.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.matches.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.matches.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.matches.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
2.親 n 個体から作成した交配家系を用いたマッピング
普通は多くの個体を同時に解析しますが、ここでは2個体のF0(AAAAA と CCCCC)から得られた2個体の F2(GGGGG と TTTTT)を解析する場合を例に説明します。
2-1. process_radtags(上の 1-1 と同じです):シーケンスデータを個体ごと(RAD タグごと)のファイルに分け、フォルダ samples に保存します。
|
tutorial username$ /Applications/stacks-xxxxx/process_radtags -p ./raw/ -o ./samples/ -b ./barcodes -c -r -e ecoRI -t 75 -i fastq |
-c オプションを指定すると、uncalled base (つまり "N")の含まれたリードが除外されます。
-e オプションでは、RAD sequencing に使った制限酵素(sbfI, pstI, ecoRI, sgrAI のいずれか)を指定します。
-t オプションでは、解析に用いる配列の長さを指定します。生データが 100 bp であっても、80 塩基目よりあとの信頼性が低い場合、81〜100 塩基目を解析から除外することができます。そのときは、頭のバーコード(今回の例では 5 bp)を除いた 6〜80 塩基目の 75 bp について解析することから、-t 75 と指定します。
-i オプションでは、データファイルの保存形式を指定します(fastq または bustard)。
他のオプションについては、Stacks のホームページにある process_radtags の説明を参照してください。
注) 久しぶりに最新のバージョン(ver 1.35)をダウンロードしてみたら、どんな拡張子でも解析してくれる訳ではありませんでした。少なくとも .fq と .fastq では解析してくれましたが、他の拡張子は試していません。(2015/10/02追記)
Tips)このパイプラインでは、フォルダ raw 内の全てのファイルを解析します。このため、フォルダ raw に別のファイルがあると、解析が上手く行きません。例えば Mac では、フォルダ raw に .DS_Store という隠しファイルが入っていることがあります。そうすると、下のようなエラーが出て解析できません。この場合は、コマンド rm ./raw/.DS_Store を実行して .DS_Store を削除してから解析して下さい。
|
Found 4 input file(s). Loaded 4, 5bp barcodes. Processing file 1 of 4 [.DS_Store] Unable to allocate Seq object. |
Tips)解析が上手く行かない場合は、データファイルを一つにまとめて、-f オプションを使ってファイル名を明示的に指定すると上手く行くことがあります。下の例では、data01.fastq, data02.fastq, data03.fastq という3つのデータファイルをひとつにまとめて、data.fastq というファイルにして解析しています。
|
(データファイルを一つにするコマンド↓) tutorial username$ cat raw/data01.fastq raw/data02.fastq raw/data03.fastq > raw/data.fastq (解析を実行するコマンド↓) tutorial username$ /Applications/stacks-xxxxx/process_radtags -f ./raw/data.fastq -o ./samples/ -b ./barcodes -c -r -e ecoRI -t 75 -i fastq |
確認)ここでは、フォルダ samples に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls samples process_radtags.log sample_AAAAA.fq sample_CCCCC.fq sample_GGGGG.fq sample_TTTTT.fq |
2-2. ustacks(オプションの説明以外は、上の 1-2 と同じです):個体ごとに、全く同じ配列を持ったリードを束ねて stack とします。また stacks 同士を比較して、個体内で同じ遺伝子座(RAD locus)のものをひとまとめにし、SNPs を検出します。今回の例では、ヘテロの SNPs (エラーによる、アリルが3つ以上の SNPs も)を検出することになります。
|
tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_AAAAA.fq -o ./stacks -i 1 -d -r -m 3 -M 2 -p 4 tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_CCCCC.fq -o ./stacks -i 2 -d -r -m 3 -M 2 -p 4 tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_GGGGG.fq -o ./stacks -i 3 -d -r -m 3 -M 2 -p 4 tutorial username$ /Applications/stacks-xxxxx/ustacks -t fastq -f ./samples/sample_TTTTT.fq -o ./stacks -i 4 -d -r -m 3 -M 2 -p 4 |
-i オプションには、個体ごとに異なる値を指定します。
-m オプションは、stack として認めるのに必要なリードの本数を指定します。個体内では、全く同じ配列を持ったリードのグループが多数存在します。このうち、リードの本数が -m の値以上のグループを stack と呼び、解析に供します。いっぽうリードが 1〜2 本しか存在しないマイナーなグループは、エラーによって生成された可能性があります。このようなエラーの影響を小さくするため、リードの本数が -m の値より小さいマイナーなグループは stack とはせず、解析から除外する事もできます。一般にはこのオプションを 3 に設定することが多いようです。しかし、coverage depth(遺伝子座あたりのリード数、下の 「Coverage depth」参照)が非常に大きい場合は、エラーによって全く同じリードが3本以上生成される確率も高まります。このことから、coverage depth が大きい個体では、-m の値を大きくするべきかも知れません。
-M オプションは、個体内で stacks が同じ遺伝子座のものと判断される際に許される塩基の違いです。つまり、2つの stacks 間の距離(異なる塩基の数)が -M の値より小さければ、両者は同じ遺伝子座に分類されます。逆に 2 つの stacks 間の距離が -M の値より大きければ、両者は異なる遺伝子座に分類されます。このため、-M の値を小さく設定しすぎると、本当は同じ遺伝子座の stacks なのに、誤って違う遺伝子座に分類してしまうリスクが高まります。逆にこの値を大きく設定しすぎると、本当は異なる遺伝子座の stacks なのに、誤って同じ遺伝子座に分類してしまうリスクが高まります。このため、適度な値を設定する必要があります。一般には、解析する個体内の遺伝的多様性が高い場合(ヘテロの遺伝子座が多い場合)には大きく、低い場合には小さく設定するのが良いでしょう。例えば F0 個体が純系であれば、遺伝的多様性が低くほとんどの遺伝子座がホモであると考えられるので、この個体については -M の値を 0 または 1 程度にして良いでしょう。F0 個体が野生個体であれば、2 もしくは 3 くらいに設定することが多いようです。F2 個体では、F0 同士が近縁であれば低めに、遠縁であれば高めに設定するのが良いと考えられます。
他のオプションについては、Stacks のホームページにある ustacks の説明を参照してください。
Tips)ustacks の複数のコマンド(上の例では AAAAA、CCCCC、GGGGG、TTTTT のコマンド)を一度に入力して解析することが出来ます。この場合、別のソフトでコマンドを作成してコピペすると便利です。しかし、改行が入っていると解析が上手く行きません。コマンドは改行ではなく、セミコロン ; でつなげると上手くいきます。
Tips)コマンドを実行すると、文字がずらずらと出力されます。その中に Mean coverage depth と Mean merged coverage depth が表示されます(両者の違いは Catchen et al.[8]をご覧下さい )。これらは、解析結果の信頼性を評価するのに重要な値です。出力を command + s (Mac の場合)で保存しておくと、あとあと便利です。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks sample_AAAAA.alleles.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
2-3. cstacks:親個体(F0 個体)のみを使って SNPs を探索します。この交配家系に存在するアリルの全ては F0 に由来するので、F0 を解析するだけで全ての SNPs を見つけることが出来ます。
|
tutorial username$ /Applications/stacks-xxxxx/cstacks -b 1 -o ./stacks -s ./stacks/sample_AAAAA -s ./stacks/sample_CCCCC -p 4 -n 3 |
-n オプションは、個体間で許されるミスマッチの数を指定します。例えば前の解析(ustacks)において、F0 オス で stack A と stack B が遺伝子座 1 に、F0 メス で stack C と stack D が遺伝子座 2 に分類されたとします。そして stack A と stack C の距離(異なる塩基の数)を A-C、ほかも同様に A-D、B-C、B-D としたとき、この4つの距離のうち最も小さい値(仮に D とします)が -n 以下であれば、遺伝子座 1 と遺伝子座 2 が相同であると判断されます。逆に D が -n より大きければ、遺伝子座 1 と遺伝子座 2 は異なるものであると判断されます。このため -n が小さすぎると、遺伝子座 1 と遺伝子座 2 が本当は相同であっても、誤って異なる遺伝子座に分類されるリスクが高まります。逆にこの値が大きすぎると、遺伝子座 1 と遺伝子座 2 が本当は異なるのに、誤って相同な遺伝子座であると分類されるリスクが高まります。よって、適切な値を設定する必要があります。一般に F0 の2個体から SNPs のカタログを作る場合、両者が近縁であれば低めに、遠縁であれば高めに設定するのが良いと考えられます。
他のオプションについては、Stacks のホームページにある cstacks の説明を参照してください。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks batch_1.catalog.alleles.tsv batch_1.catalog.snps.tsv batch_1.catalog.tags.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
2-4. sstacks(上の 1-4 と同じです):各個体で、SNPs の遺伝子型を決めます。
|
tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_AAAAA -o ./stacks -p 4 tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_CCCCC -o ./stacks -p 4 tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_GGGGG -o ./stacks -p 4 tutorial username$ /Applications/stacks-xxxxx/sstacks -b 1 -c ./stacks/batch_1 -s ./stacks/sample_TTTTT -o ./stacks -p 4 |
オプションについては、Stacks のホームページにある sstacks の説明を参照してください。
注)Stacks ホームページの FAQ では、ここで progeny(本ホームページの例では F2 個体)のみを解析するような書き方をしています。しかしそうすると、次の genotypes で joimap などの入力ファイルを作成する際に、親個体の遺伝子型を考慮してくれません。F0 個体も解析に加えるべきでしょう。
Tips)sstacks の複数のコマンド(上の例では AAAAA、CCCCC、GGGGG、TTTTT のコマンド)を一度に入力して解析することが出来ます。この場合、別のソフトでコマンドを作成してコピペすると便利です。しかし、改行が入っていると解析が上手く行きません。コマンドは改行ではなく、セミコロン ; でつなげると上手くいきます。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks batch_1.catalog.alleles.tsv batch_1.catalog.snps.tsv batch_1.catalog.tags.tsv sample_AAAAA.alleles.tsv sample_AAAAA.matches.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.matches.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.matches.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.matches.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
2-5. genotypes:joinmap などの入力ファイルを作ります。
|
tutorial username$ /Applications/stacks-xxxxx/genotypes -b 1 -P ./stacks -t F2 -c -o joinmap -r 1 |
-t オプションでは、マップのタイプを指定します(CP, DH, F2, BC1, GEN)。
-o オプションでは、 joinmap, onemap, rqtl, genomic で読み込むファイルを作成します。
ほかのオプションについては、Stacks のホームページにある genotypes の説明をご覧ください。
Tips) joinmap などの入力ファイルを作成する際、cstacks でカタログを作成するのに用いた個体を「親(本ホームページの例では F0)」とし、それ以外の個体をマッピングに用いる個体(同 F2)として解析します。よって、cstacks でどの個体を解析に使ったかによって、結果が異なります(例えば cstacks で全個体を解析に使った場合、マッピングに用いる個体が無くなってしまいます)。また、sstacks で解析した個体によっても結果が異なります(「出力ファイルの見方」も参照)。cstacks、sstacks に用いる個体は、交配実験のデザインや行いたい解析によって適切に決める必要があるでしょう。
確認)ここでは、フォルダ stacks に下のファイルが作られます(黄色文字)。
|
tutorial username$ ls stacks batch_1.catalog.alleles.tsv batch_1.catalog.snps.tsv batch_1.catalog.tags.tsv batch_1.genotypes_1.loc batch_1.haplotypes_1.tsv sample_AAAAA.alleles.tsv sample_AAAAA.snps.tsv sample_AAAAA.tags.tsv sample_CCCCC.alleles.tsv sample_CCCCC.snps.tsv sample_CCCCC.tags.tsv sample_GGGGG.alleles.tsv sample_GGGGG.matches.tsv sample_GGGGG.snps.tsv sample_GGGGG.tags.tsv sample_TTTTT.alleles.tsv sample_TTTTT.matches.tsv sample_TTTTT.snps.tsv sample_TTTTT.tags.tsv |
出力ファイルの見かた
主要な4つのファイルについて説明します。ファイルサイズが大きく Excel などで開くのが難しい場合は、カレントディレクトリを stacks にしてから「less batch_1.haplotypes_1.tsv」などと入力して実行すると、ファイルの最初の方から少しずつ見ることが出来ます。less から抜け出すには、q を押します。
1.batch_1.haplotypes_1.tsv
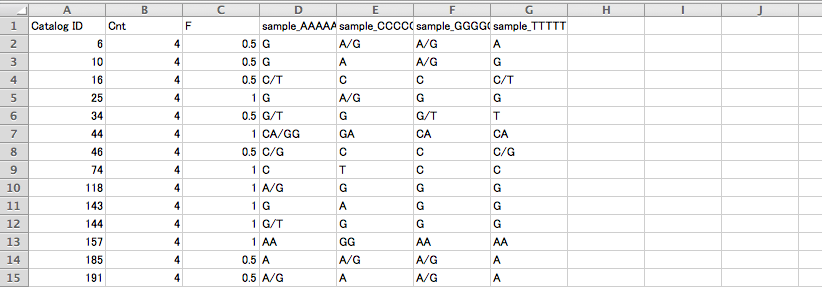
各遺伝子座(RAD locus)の遺伝子型の表です。A列は各遺伝子座の Catalog IDです。例えば Catalog #6 の遺伝子座には A/G という SNP があり、左の個体から G (D列), A/G (E列), A/G (F列), A (G列) という遺伝子型が検出されたことを示しています。
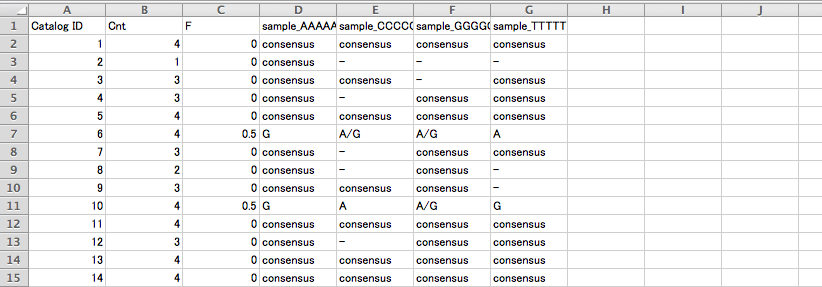
consensus は、その遺伝子座には SNP が見つからなかったという意味です。このような遺伝子座を除いて SNPs のある遺伝子座だけのファイル(hoge.tsv)を作るには、カレントディレクトリを stacks にしてから「grep -v "consensus" batch_1.haplotypes_1.tsv > hoge.tsv」と入力して実行します。
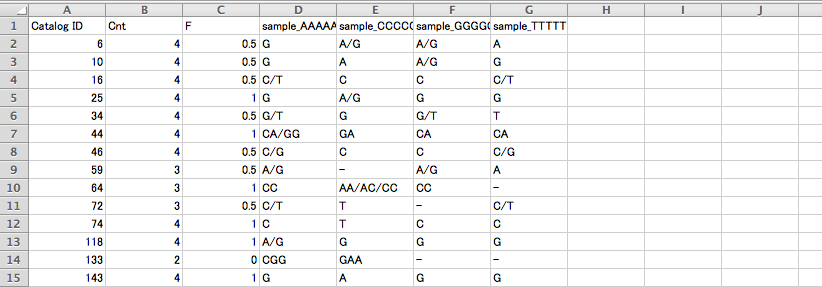
-はヌル(シーケンスが読まれなかった)です。ヌルのある遺伝子座を除いたファイル(hogehoge.tsv) を作るには、カレントディレクトリを stacks にしてから「grep -v "-" hoge.tsv > hogehoge.tsv」と入力して実行します。
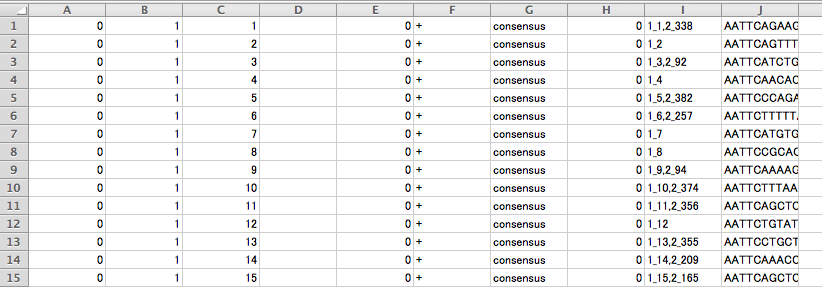
2.batch_1.catalog.tags.tsv
各遺伝子座の配列を確認できます。情報の表示される列は、Stacks のバージョンによって多少異なります。C列の数字は batch_1.haplotypes_1.tsv の Catalog ID に対応します。
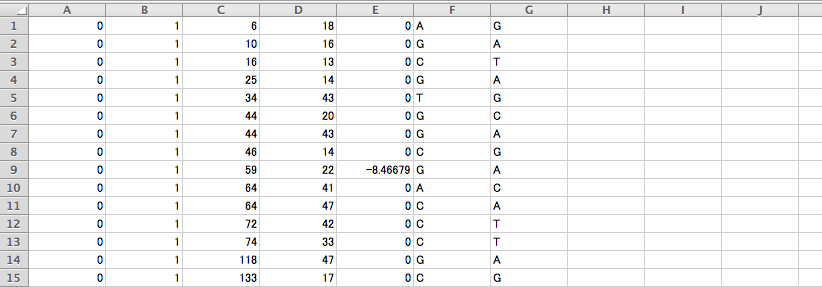
3.batch_1.catalog.snps.tsv
SNP の位置の表です。例えば下図の1行目は、Catalog #6(C 列) の遺伝子座の 18 番目の塩基(D 列、最初の塩基を 0 と数える)に A/G の SNP(F 列とG 列) があることを示しています。
4.batch_1.genotypes_1.loc
joinmap などの入力ファイルです。パイプライン genotypes で -o オプションに joinmap などを指定すると作成されます。このファイルの説明を Stacks のホームページなどで見つけることが出来ませんでしたが、他のファイルと見比べたところ、以下のようになっているようです。間違いがあるかも知れませんが、参考まで。
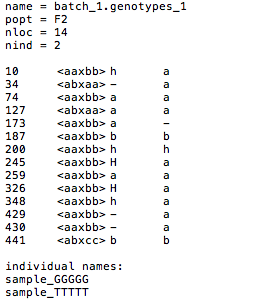
上の図は、genotypes のオプションで -t F2 -c -o joinmap と指定した場合の出力です。左端の数字(10, 34, 74, ...)は、batch_1.haplotypes_1.tsv の Catalog ID に対応します。出力されている遺伝子座は、以下の条件を満たしたものだけのようです:F0 の2個体を a, b としたとき、F2 が a からだけ(下の表の遺伝子座 1 と 5)、b からだけ(同 2)、もしくは a, b の両方から(同 3)アリルを受け継いだと特定できる遺伝子座。これらの遺伝子座における F2 の遺伝子型は、上の図でそれぞれ a, b, h と出力されています(右の2列、<> 内のアルファベットとは対応しません)。F2 の遺伝子型が a, b, h のいずれにも特定できない場合(下の表の遺伝子座 4, 6-8)は - と出力されます。大文字(上の図の H)は、genotypes のオプション -c を指定した際に修復された遺伝子型を示しているようです。
| F0-a の遺伝子型 | F0-b の遺伝子型 | F2 の遺伝子型 | 出力 | 出力の理由 | |
| 遺伝子座 1 | A/A | G/G | A/A | <aaxbb> a | a 以外あり得ない |
| 遺伝子座 2 | A/A | G/G | G/G | <aaxbb> b | b 以外あり得ない |
| 遺伝子座 3 | A/A | G/G | A/G | <aaxbb> h | h 以外あり得ない |
| 遺伝子座 4 | A/A | G/G | - | <aaxbb> - | 情報なし |
| 遺伝子座 5 | A/G | G/G | A/A | <abxaa> a | a 以外あり得ない |
| 遺伝子座 6 | A/G | G/G | G/G | <abxaa> - | a, b, h の可能性あり |
| 遺伝子座 7 | A/G | G/G | A/G | <abxaa> - | a, h の可能性あり |
| 遺伝子座 8 | A/G | G/G | - | <abxaa> - | 情報なし |
並列処理
パイプライン ustacks, cstacks, sstacks では、オプション -p によって使用するCPUの数を変えられます。この他にも、UNIXのウィンドウを複数開いて、複数のコマンドを同時に実行することによって、簡単に並列処理をすることができます。
Coverage depth
Coverage depth は個体ごと、遺伝子座ごとのリード数で、データの信頼性を評価するのにとても重要な値です。この値は、一般には個体ごとの平均値として表されます。以下に示すように、この値は高いときよりも、低いときのリスクの方が深刻です。
Coverage depth が低いときのリスク
この値が小さい個体では、遺伝子型が正確に再現されない可能性が高まります。例えば、ある遺伝子座で本当は A/G なのに、アリル G を示すリードが少なく解析から除外されると(もしくはアリル G のリードが全く無ければ)、誤って A/A と判定されてしまいます。さらにアリル A を示すリードも少なければ(もしくは無ければ)、誤ってヌルと判定されてしまいます。このような誤判定は確率的に生じ、coverage depth の低い個体ではその確率が高まります。つまり、他の個体より見かけ上のホモやヌルの遺伝子座が多くなります。
Coverage depth が高いときのリスク
いっぽうこの値が高い個体では、リード数が多いため、その分エラーの数も多くなります。そうすると、誤った配列をもつ「間違いリード」も多くなり、偶然にも全く同じ配列をもつ「間違いリード」が何本も生じる可能性が高まります。全く同じ「間違いリード」がある本数以上(ustacks の -m の値以上)あると、stack として認識され、解析に影響を与えてしまいます。しかし、そのような「間違いリード」の多くは、ustacks の -m の値を大きくすることによって、解析から除外することが出来ます。このため、このリスクはそれほど深刻ではないと考えられます。
Coverage depth の調節
以上のように、coverage depth は高い方が良いと考えられます。適正な値は一概には言えませんが、私の経験では、リファレンスゲノムが無い場合で 30 以上ではないかと感じています。Coverage depth は、使用する制限酵素、リード数、解析に供する個体数などの影響を受けます(下の式を参照)。しかし、今回ご紹介したデータ解析の段階では、基本的には coverage depth を上げることはできません(stacks に振り分けられなかったマイナーなリードを利用して coverage depth を上げる方法は Catchen et al.[8]を参照してください)。このため、十分な coverage depth が得られるよう、事前の実験計画が重要となります。実験計画を立てるには、RAD のホームページにある RAD counter が便利です。
coverage depth = リード数 / 個体数 / 遺伝子座数
遺伝子座数 = ゲノムサイズ x 制限領域の頻度 x 2
制限領域の頻度 = (GC / 2)a x (0.5 - GC / 2)b
GC: ゲノム中の GC の割合
a: 制限酵素の認識配列中の GC の数(SbfI の認識配列 CCTGCAGG では6)
b: 制限酵素の認識配列中の AT の数(SbfI の認識配列 CCTGCAGG では2)
Coverage depth の調べ方
ustacks の説明のところでも書きましたが、coverage depth を調べるには、ustacks を実行したときの出力を見ます(出力は、Mac の場合コントロール + s で保存できます)。RAD タグごとに、Mean coverage depth と Mean merged coverage depth の2つがあります(2つの違いは Catchen et al.[8]を参照してください)。Stacks のマニュアルに従って denovo_map.pl を実行した場合は(AMP 環境が必要)、フォルダ stacks に denovo_map.log というファイルが保存され、そこに coverage depth が出力されます。
最後に
ここでは、MySQL を使わずに Stacks を走らせる方法を紹介しました。念のため、小さなデータセット(25000 リード×4個体)を使って MySQL を使った場合と使わなかった場合で結果を比較しました(テストには Stacks-0.998 を使用)。その結果、集団解析、マッピングのどちらにおいても batch_1.haplotypes_1.tsv の出力に違いのないことを確認しました。
MySQL を使った場合は、結果を PHP を使ってブラウザ上に出力したり、Spreadsheet-WriteExcel を使って Excel ファイルに出力したりすることができます。これらの出力では、上で紹介した出力ファイル(batch_1.haplotypes_1.tsv, batch_1.catalog.tags.tsv, batch_1.catalog.snps.tsv)が統合されて見やすくなります。しかし、今回ご紹介した方法ではこれらの機能を使うことはできません。そのかわり、MySQL を介さずにこれらの出力ファイルを統合する perl スクリプトを作りました(script2.txt.zip, 4 KB)。解凍して出力ファイルと同じフォルダ(上の例では stacks)に入れ、ターミナル上でカレントディレクトリをそのフォルダにし、「perl script2.txt」と入力して実行します。あとは指示通りに操作します。分からないことがありましたら、ご連絡下さい(メール)。
注)「Can't exec /usr/bin/perl at script2.txt」というエラーメッセージが出ることがあります。 その原因として、perl が /usr/bin/ に入っていない場合があります。 この場合は、まずターミナルでコマンド which perl を実行して、perl のディレクトリを確認します。 次に script2.txt を mi などのテキストエディタで開き、一行目 #!/usr/bin/perl の「/usr/bin/perl」の部分を、先ほど調べた perl のディレクトリに書き換えて上書き保存します。 改行は必ず LF (UNIX) にして下さい。
batch_1.haplotypes_1.tsv から NEXUS ファイルを作る perl スクリプトはこちらです(MultiSNPs2.txt.zip, 4KB)。使い方は script2.pl.zip と同じです(ただし、実行コマンドは「perl MultiSNPs2.txt」)。不明な点がありましたら、ご連絡下さい(メール)。
この小文が皆さんの研究の一助になれば幸いです。
謝辞 当ホームページを作成するにあたり、さまざまな方々の協力を得ました。この場を借りてお礼申し上げます。
引用
ホームに戻る